The p53 Tumor Suppressor Gene
The most frequently mutated gene in human cancers—occurring in more than 50 percent of all cancers—is the p53 gene. The p53 gene encodes a nuclear protein that acts as a transcription factor that represses or stimulates transcription of more than 40 different genes. (A transcription factor is a regulatory protein that binds to DNA sequences and affects transcription of specific genes.)
Normally, the p53 protein is continuously synthesized but is rapidly degraded and therefore is present in cells at a low level. In addition, the p53 protein is normally bound to another protein called Mdm2, which prevents the phosphorylations and acetylations that convert the p53 protein from an inactive to an active form. Several types of events cause a rapid increase in the levels of activated p53 protein. These include chemical damage to DNA, double-stranded breaks in DNA induced by ionizing radiation, or the presence of DNA-repair intermediates generated by exposure of cells to ultraviolet light. The increase in activated p53 protein results from increases in protein phosphorylation, acetylation, and an increase in p53 protein stability.
The p53 protein initiates two different responses to DNA damage: arrest of the cell cycle followed by DNA repair, or apoptosis and cell death if DNA cannot be repaired. Each of these responses is accomplished by p53 acting as a transcription factor that stimulates or represses gene expression.
In normal cells, p53 can arrest the cell cycle at several phases. To arrest the cell cycle at the G1/S checkpoint, activated p53 protein stimulates transcription of a gene encoding the p21 gene product. The p21 protein inhibits the CDK4/cyclin D1 complex, preventing the cell from moving from G1 phase into S phase. Activated p53 protein also regulates expression of genes that retard the progress of DNA replication, allowing time for DNA damage to be repaired during S phase. By regulating expression of other genes, activated p53 can block cells at the G2/M checkpoint, if DNA damage occurs during S phase.
Activated p53 can also instruct a damaged cell to commit suicide by apoptosis. It does so by activating the transcription of the Bax gene and repressing transcription of the Bc12 gene. In normal cells, the BAX protein is present in a heterodimer with the Bc12 protein and the cell remains viable. When the levels of BAX protein increase in response to p53 stimulation of Bax gene transcription, BAX homodimers are formed and these homodimers activate the cellular changes that lead to cellular self-destruction.
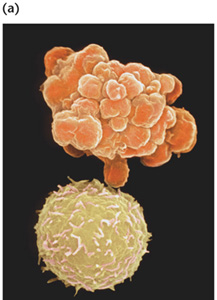
(a)
A normal white blood cell (bottom) and a WBC undergoing apoptosis. Apoptotic
bodies appear as grape-like clusters on the cell surface (b) The relative
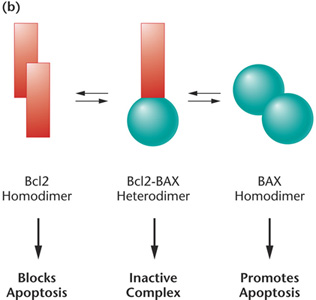
concentrations of the Bc12 and BAX proteins regulate apoptosis. A normal
cell contains a balance of Bc12 and BAX, which form inactive heterodimers
(center). A relative excess of Bc12 results in Bc12 homodimers, which
prevent apoptosis (left). Cancer cells with Bc12 overexpression are resistant
to chemotherapies and radiation therapies. A relative excess of BAX results
in BAX homodimers, which induce apoptosis (right). In normal cells, activated
p53 protein induces transcription of BAX and inhibits transcription of
Bc12, leading to cell death. In many cancer cells, p53 is defective, preventing
the apoptotic pathway from removing the cancer cells. |
 |
 |
In cancer cells that lack functional p53, BAX protein levels do not increase in response to cellular damage and apoptosis may not occur. Consequently, cells lacking functional p53 are unable to arrest at cell cycle checkpoints or to enter apoptosis in response to DNA damage. As a result, they move unchecked through the cell cycle, regardless of the condition of the cell's DNA. Cells lacking p53 have high mutation rates and accumulate the types of mutations and chromosomal aberrations that lead to cancer. Because of the importance of the p53 gene to genomic integrity, it is often referred to as the "guardian of the genome".